-

Infection, Immunity & Inflammation
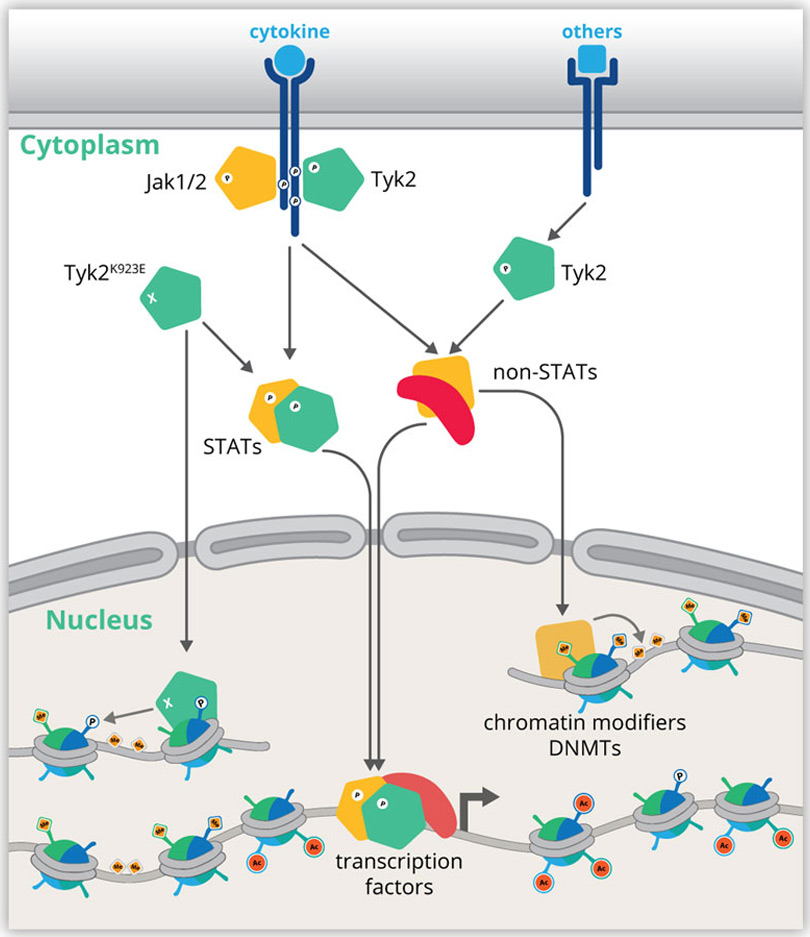
In the early 90ies of the last century experiments on the cellular response mechanisms of interferons led to the identification of the JAK-STAT pathway as a paradigm for signaling from the cell surface to chromatin. JAK-STAT controls the development and activity of hematopoietic cells. They provide inflammatory and immune stimuli and thereby regulate host-pathogen interactions, tumor surveillance and inflammatory processes.
MORE
-

Hematopoietic Malignancies
Constitutive JAK and/or STAT activation is found in most myeloproliferative neoplasms and in many other hematopoietic malignancies. It is frequently a biomarker of poor prognosis. The underlying molecular aberrations include activating mutations in or overexpression of signaling components (cytokine receptor, JAK, STAT), or rare JAK fusion proteins.
MORE
-

Homeostatic Cell Type-Specific Regulation
Cellular and tissue homeostasis is the result of variables being regulated on chromatin level such that the internal environment remains stable and fairly constant even though the external environment varies. Cell type-specific homeostatic chromatin landscapes are maintained by tonic (constitutive) signals
provided by auto- and paracrine acting cytokines and growth factors as well as by cell-cell contacts. All SFB members contribute to the generation of a map of the JAK-STAT-shaped chromatin landscape of various hematopoietic and structural cell types under homeostatic in comparison to stressed or diseased conditions.
MORE
-

The Epigenome of JAK-STAT
Recently molecular genetics research focused on global aspects of gene control and the underlying hierarchies. Our proposed projects aim to place the interactomes of JAKs and STATs into the emerging global chromatin landscape. Genome-wide analyses integrate the various layers of RNA, DNA and protein dynamics to pinpoint key regulators of pathways and pathway hierarchies that drive or prevent disease. We aim to determine the impact of JAKs and STATs on 3D genome architecture, landscapes of chromatin modifications, DNA methylation and transcription factor binding.











{kind=link}